We model the interaction of light and matter as well as chemical reactions in real time. Our aim is not only to unravel the underlying processes but also to control them. The systems under consideration reach from diatomics to DNA. Accordingly, different techniques like quantum dynamics or molecular dynamics calculations are employed.
Neural networks and machine learning
Artificial neural networks are computer algorithms mimicking the operating mode of our brain. These neural networks belong to the vast field of machine learning and have been employed for a wide range of applications. We use them to predict potential energy surfaces and other molecular properties. These neural networks offer the exciting advantage of delivering highly accurate results at little computational cost.
SHARC - Surface Hopping including ARbitrary Couplings
We have developed a nonadiabatic ab initio molecular dynamics (MD) method including spin-orbit coupling (SOC) and laser fields. This mixed quantum classical dynamics method is used as a general tool for studies of excited-state processes. Intersystem crossing - e.g. a transition from a singlet to a triplet state - can be investigated within the given framework. The laser interaction is treated on a non-perturbative level that allows to consider nonlinear effects like strong Stark shifts. As MD allows for the handling of many atoms, the interplay between triplet and singlet states of large molecular systems are accessible.
Ab initio molecular dynamics software SHARC: sharc-md.org
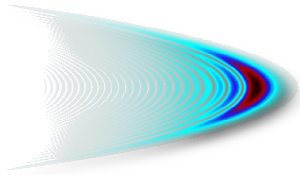
Laser control of chemical reactions
One of the central problems in chemistry is to control the outcome of reactions. This goal can be achieved using laser light. In general, every molecular system can be modified by a molecule-field interaction to yield a desired product. This universality arises from the large variety of laser parameters, which can be adapted to yield electro-magnetic fields of the most different shapes and colors (see e.g. the Wigner representation of a third-order chirped pulse on the left). In this way, chemical bonds can be broken and formed selectively. We use quantum dynamics (QD) simulations to unravel the mechanism underlying such photodissociation and association processes.