Das Molekülorbital-Modell

H2 molecule forming from two distant hydrogen atoms and corresponding evolution of orbitals.

H2 molecule antibonding orbital forming.
Link zu den Folien:
Wir simulieren die Wechselwirkung zwischen Licht und Materie sowie chemische Reaktionen in Echtzeit. Unser Ziel ist nicht nur die molekularen Prozesse zu verstehen, sondern sie auch zu kontrollieren. Die hierzu verwendeten Modellsysteme reichen von zweiatomigen Molekülen bis zu DNA in wässriger Lösung. Entsprechend unterschiedliche Methoden wie Quantendynamik oder klassische Molekulardynamik finden bei den Berechnungen Verwendung.

Neuronale Netze und maschinelles Lernen
Künstliche neuronale Netze sind Computer-Algorithmen, die die Funktionsweise unseres Gehirns imitieren. Diese neuronalen Netze gehören zum Forschungsfeld des maschinellen Lernens und werden in den unterschiedlichsten Anwendungsbereichen eingesetzt. Wir nutzen sie, um Potenzialenergieflächen und andere molekulare Eigenschaften vorherzusagen. Diese neuronalen Netze haben den überragenden Vorteil, dass sie hochgenaue Resultate bei geringem Rechenaufwand liefern.
 SHARC - Surface Hopping including ARbitrary Couplings
SHARC - Surface Hopping including ARbitrary Couplings
Wir haben eine nicht-adiabatische ab initio Molekulardynamik-Methode entwickelt, welche Spin-Bahn-Kopplung und Laser-Wechselwirkungen beschreiben kann. Diese gemischt quanten-klassische Dynamik-Methode verwenden wir zur Untersuchung von Prozessen in angeregten Zuständen. Intersystem crossing - z.B. ein Übergang von einem Singulett in einen Triplett-Zustand - kann in diesem Rahmen erforscht werden. Die Laser-Wechselwirkung wird nicht störungstheoretisch behandelt, so dass nichtlineare Prozesse, beispielsweise durch einen Stark-Effekt induziert, beobachtet werden können. Da Molekulardynamik erlaubt eine große Anzahl von Atomen zu modellieren, wird das Zusammenspiel zwischen Singulett- und Triplett-Zuständen in großen Molekülen zugänglich.
Ab initio molecular dynamics software SHARC: sharc-md.org
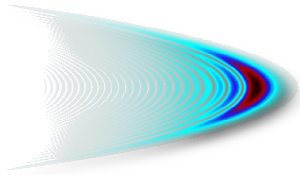
Laserkontrolle von chemischen Reaktionen
Eines der zentralen Probleme in der Chemie ist, das Ergebnis von Reaktionen zu kontrollieren. Dieses Ziel kann mittels Laserlicht erreicht werden. Im Allgemeinen lässt sich jedes molekulare System durch Molekül-Feld-Wechselwirkungen steuern und so ein gewünschtes Produkt erhalten. Diese Universalität leitet sich aus der unendlichen Vielzahl der Laserparameter ab, die so angepasst werden können, dass elektromagnetische Felder unterschiedlichster Form und Farbe erzeugt werden können (vgl. z.B. die Wigner-Verteilung eines Pulses mit Chirp dritter Ordnung in der Abbildung). Auf diese Weise können chemische Bindungen selektiv geformt und gebrochen werden. Wir verwenden Quantendynamik-Simulationen, um die Mechanismen aufzudecken, die solchen Photodissoziations- und Assoziations-Prozessen zugrundeliegen.
Falls Sie an ab-initio-Molekulardynamik interessiert sind, können Sie auch die Seite sharc-md.org (nur Englisch) besuchen.